Background
I have been collaborating with Jonathan Walton,
a professor in Plant biology department at MSU on genome assembly of a toxic mushroom called
destroying angel (A. bisporigera).
Right now, we have 9 datasets from PacBio in bas.h5 format (a movie file).
The file size is about 1.5-3.0 Gb.
Moreover, there are also files in fasta, fastq and ccs.fastq. However, I learned later that those
files were useless because they were not quality filtered and still contained adapters.
Quality Filtering and Trimming with SMRT PreAssembly Pipeline
SMRT pipeline is available on github here
and also pre-installed on EC2 machine (ami-fbbbcf92), so I launched EC2 instance and learned to run the software.
The software has several pipelines, each consists of several modules.
You can also build your own pipeline by putting the modules together.
I ran AlloraSFilter module on the command line with smrtpipe.py with quality score 0.75 to all bas.h5 files.
Here are the results.
|
Pre Filter |
Post Filter |
| Polymerase Read Bases |
1,784,846,157 bp |
1,163,287,035 bp |
| Polymerase Reads |
676,377 |
193,263 |
| Polymerase Read Length |
2639 bp |
6019 bp |
| Polymerase Read Quality |
0.301 |
0.835 |
As you can see, the polymerase read length increases from 2639 bp to 6019 bp and the mean quality score also
increases from 0.30 to 0.83. The plots below show a histogram of read lengths and quality scores of polymerase reads.
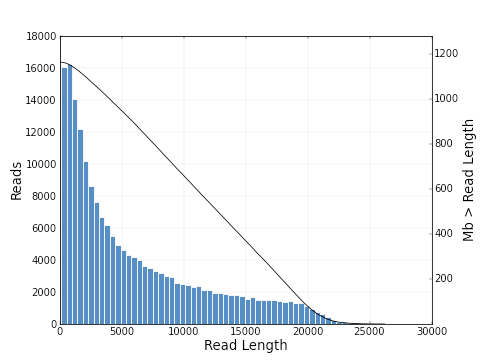
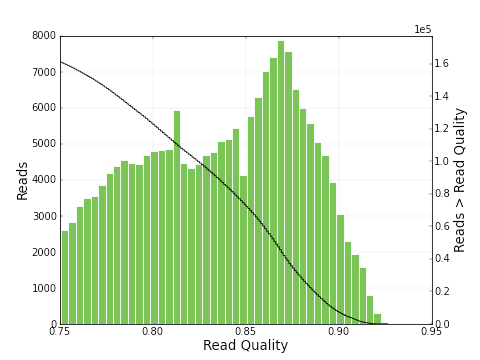
And here is a histogram of subread lengths (after adaptors are removed).
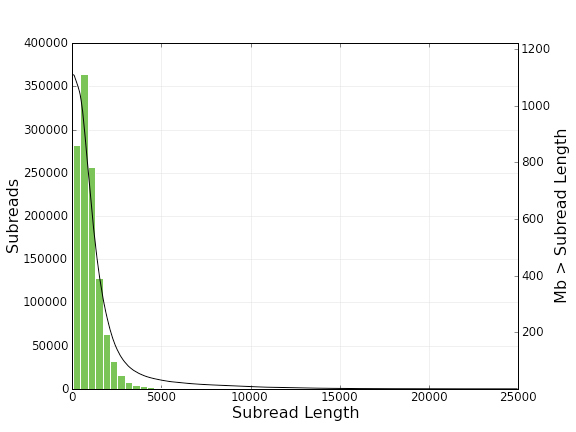
The module also generated fastq and fasta file for both subreads and consensus reads (CCS) from bas.h5 files.
Error Correction and Assembly
After that, I ran pacToCA to error correct subreads with CCS sequences and run Celera assembler to assembled corrected reads.
Celera produced five different output files, singletons, unitigs, contigs, degenerate contigs and scaffolds.
Basically, it built unique contigs from reads, and reads not used to build unique contigs were stored in singletons file.
Then unique contigs were aligned to build contigs. Unique contigs that were not aligned to any other contigs were stored in degenerate contigs file.
Therefore, the contigs file contains high confident contigs (multiple alignments) and degenerate file contains low confident contigs.
Scaffolds contain scaffolded contigs; however, in our case no contigs were scaffolded, so it contains exactly the same contigs in contigs file.
Unfortunately, the total length of high confident contigs is only 6.4 Mbp, whereas that of low confident contigs is about 8.9 Mbp.
The max contig size is 13,072 bp and the max size of degenerate contig is 78,021 bp. N50 sizes are 2,147 bp and 997 bp respectively.
I know the estimated size of the genome is ~50 Mbp, so this really low. However, I learned that the overall assembly size is
comparable to that of the assembly done by other collaborators.
So, the issue could be from DNA samples or sequencing, not from the assembly.
Also, I tried to combined 454 assembly that was done by someone with PacBio assembly from Celera assembly.
I combined singletons, unique contigs, degenerate contigs and 454 contigs and assembled them with CAP3 genome assembly.
The results show that the total contig size is much greater (17.5 Mbp) and the total size of singleton contigs is about 21 Mbp.
Max contig size also increases (24 Kbp) as well as N50 size (1.4 Kbp).
Conclusion
It seems like mushroom genome with PacBio runs into the same problem with other genome assembly projects.
A lot of reads and contigs (even with length = 78 Kbp) cannot be assembled into long contigs.
It also appears that 454 assembly also have unique contigs that cannot be merged to PacBio assembly.
These problems may be solved by paired-end with different insert sizes and Illumina sequencing.
There are comments.